Laboratory of Microbial Resources and Catalysis, School of Biological Engineering, Dalian Polytechnic University, Dalian 116033, Liaoning, China
Received: 07-12-2021; Accepted: 24-03-2022; Published online: 26-04-2022
Foundation item: National Natural Science Foundation of China (31771907)
#These authors equally contributed to this work
异麦芽酮糖俗称棕榈糖是由D-葡萄糖和D-果糖通过α-1, 6-糖苷键连接而成的蔗糖的结构异构体[1-2]。异麦芽酮糖具有与蔗糖相似的物理和感官特性,但又具有优于蔗糖的生理功能,如异麦芽酮糖是非致龋性的且消化速度慢、能量释放时间长、不会显著提高胰岛素水平及血糖指数等,因此被广泛用作功能性甜味剂[1-4]。由于异麦芽酮糖在工业中具有广泛的应用前景,寻求一种高效经济的方式来生产异麦芽酮糖是人们一直以来的研究热点[3]。目前,异麦芽酮糖的生产主要是通过蔗糖异构酶(EC5.4.99.11)转化蔗糖实现。利用蔗糖异构酶转化蔗糖生产异麦芽酮糖现阶段主要有2种方式:(1) 利用微生物异源表达的蔗糖异构酶转化蔗糖,例如利用枯草芽孢杆菌(Bacillus subtilis) WB800表达来自克雷伯氏菌(Klebsiella singaporensis) LX3的蔗糖异构酶转化蔗糖产生异麦芽酮糖[5],利用大肠杆菌(Escherichia coli) BL21/pET22b-sim表达土壤分散泛菌(Pantoea dispersa) UQ68J的蔗糖异构酶(PdSIase)转化蔗糖产生异麦芽酮糖[6]等;(2) 采用固定化技术将蔗糖异构酶进行固定化后再转化蔗糖产生异麦芽酮糖。酶的固定化技术分为2种:(1) 通过物理吸附或者材料包埋的方式将酶进行固定化,例如以硅藻土吸附法与微胶囊包埋法2种方法对欧文氏菌(Erwinia sp.) D12来源的蔗糖异构酶进行固定化[7];(2) 细胞固定化,主要通过细胞表面展示(cell-surface display)技术完成,例如利用解脂耶氏酵母(Yarrowia lipolytica)展示来自P. dispersa UQ68J的蔗糖异构酶等[3]。
细胞表面展示是一种经济、高效的蛋白质工程技术,其通过将目的蛋白的编码基因与合适的蛋白锚定基序融合,可以在单个宿主菌中完成基因的表达、酶的固定化及蛋白质的纯化,从而降低蛋白纯化所需成本[8]。根据表达宿主的不同,锚定蛋白的选择也是多种多样的,例如:在B. subtilis表面利用外孢子皮蛋白(CotX)展示L-阿拉伯糖异构酶及蔗糖异构酶[9]等;在酿酒酵母(Saccharomyces cerevisiae) BY4741中通过3种不同的锚定蛋白(Sag1p、Sed1p和Cwp2p)展示β-葡萄糖苷酶[10];在Y. lipolytica中通过一段功能未知的蛋白(YALI0F24255p)在细胞表面展示大豆表皮过氧化氢酶[11]。
解脂耶氏酵母(Y. lipolytica)是公认的适合表面展示的菌株之一,被归类为“普遍公认的安全(generally recognized as safe,GRAS)”微生物。其拥有清晰的遗传背景、易于培养和生产成本低廉等特点,可用于从高浓度糖中生产多种食品化合物,例如有机酸及脂类等[12-14]。据报道,目前已成功在Y. lipolytica中展示了α-半乳糖苷酶Ⅱ[15]、脂肪酶[16]、β-内酰胺酶[17]、β-葡萄糖苷酶[9]、L-左旋天冬酰胺酶[18]、蔗糖异构酶等,其中蔗糖异构酶却只成功展示了来自P. dispersa UQ68J的蔗糖异构酶,其他来源的并未研究,并且表面展示的蔗糖异构酶仅在20−40 ℃的温度及pH 4.0−7.0区间内较稳定[2]。
本研究以Y. lipolytica Po1g为表达菌株,利用Y. lipolytica细胞壁锚定蛋白Pir1构建表面展示重组菌,以此将源自Klebsiella sp. LX3的蔗糖异构酶PalI展示在Y. lipolytica Po1g表面。通过对重组菌体展示的蔗糖异构酶催化蔗糖底物产生的产物含量的分析,探索表面展示系统对产物生成的影响及工业上通过转化蔗糖生成异麦芽酮糖减少副产物单糖的应用。
1 材料与方法
1.1 菌株及载体
克隆菌株大肠杆菌(E. coli) DH10B,大连工业大学生物催化技术国家地方联合工程实验室;展示菌株Y. lipolytica Po1g及载体pYLSC,中国台湾益生生技有限公司;克隆载体T-vector pMD19 (simple),宝生物工程(大连)有限公司;携带Y. lipolytica细胞壁锚定蛋白Pir1编码基因的质粒pUC57-Pir1由生工生物工程(上海)股份有限公司全基因合成;pYLSC-PalI质粒由大连工业大学生物催化技术国家地方联合工程实验室构建并保存。
1.2 培养基
YPD液体培养基(pH 4.0)用于解脂耶氏酵母的发酵培养,YPD培养基使用前按1:10加入0.5 mol/L柠檬酸-柠檬酸钠缓冲液;LB液体培养基(pH 7.0)用于大肠杆菌的发酵培养;YNB固体培养基用于解脂耶氏酵母转化子的筛选[11]。
1.3 主要试剂
TaKaRa rTaq™ DNA Polymerase、PrimerSTAR® HS DNA Polymerase、QuickCut™ Xba I、QuickCut™ Sfi I、QuickCut™ Dpn I、T4 DNA Ligase、Protein Molecular Weight Marker (High,Low)、DNA片段纯化试剂盒、质粒DNA小量纯化试剂盒、DNA凝胶回收试剂盒,宝生物工程(大连)有限公司;YLEX Expression Kit,中国台湾益生生技有限公司;胰蛋白胨、无水葡萄糖、蔗糖、琼脂粉和Goldview Ⅰ核酸染料,北京鼎国昌盛有限责任公司。
解脂耶氏酵母的培养需添加0.5 mol/L柠檬酸-柠檬酸钠缓冲液(pH 4.0);表面展示的蔗糖异构酶酶活力的测定采用0.1 mol/L磷酸氢二钠-柠檬酸缓冲液(pH 6.0);表面展示的蔗糖异构酶最适pH及pH稳定性测定采用0.1 mol/L磷酸氢二钠-柠檬酸(pH 3.0−6.0)、0.2 mol/L磷酸氢二钠-磷酸二氢钠(pH 6.0−8.0)和0.05 mol/L Tris-HCl (pH 8.0−9.0)。
1.4 引物设计及合成
以全基因合成的锚定蛋白Pir1编码基因Pir1序列为模板,设计扩增锚定蛋白Pir1编码基因Pir1的引物YlP-SI-SOE1-F和YlP-SI- SOE1-R;以pYLSC-PalI质粒为模板,设计扩增蔗糖异构酶编码基因PalI的引物YlP-SI- SOE2-F和pYLSC-SI-R。PCR反应体系(25 μL):正、反向引物(10 μmol/L)各1 μL,ddH2O 14.75 μL,模板基因(100 μg/μL) 1 μL,5×PrimerSTAR Buffer (Mg2+ Plus) 5 μL,PrimerSTAR® HS DNA Polymerase (2.5 U/μL) 0.25 μL,dNTP Mixture (各2.5 mmol/L) 2 μL。PCR反应条件:95 ℃ 5 min;94 ℃ 45 s,55 ℃ 45 s,72 ℃ 2 min,共35个循环;72 ℃ 10 min。通过2个基因重叠的部分进行重叠延伸PCR,获得融合基因。PCR反应体系(25 μL):Pir1基因片段(100 ng/μL)与PalI基因片段(200 ng/μL)各1 μL,ddH2O 15.75 μL,5×PrimerSTAR Buffer (Mg2+ Plus) 5 μL,PrimerSTAR® HS DNA Polymerase (2.5 U/μL) 0.25 μL,dNTP Mixture (各2.5 mmol/L) 2 μL。PCR反应条件:95 ℃ 5 min;98 ℃ 30 s,68 ℃ 2 min,共15个循环;68 ℃ 10 min。引物PMD-P1和PMD-P2为T-vector pMD19 (simple)亚克隆载体鉴定的通用引物,引物6560F和6904R为鉴定表面展示载体的通用引物。实验中用到的所有引物序列如表 1所示,均由生工生物工程(上海)股份有限公司合成。
表 1 本文中使用的引物
Table 1 Primers used in this study
| Peimer name |
Peimer sequence (5′→3′) |
Primer description |
| YlP-SI-SOE1-F |
CGGCCGTTCTGGCCATGCTCTTCAAGTCCGCTGCC |
Amplification of Pir1 gene |
| YlP-SI-SOE1-R |
ctgattcaaggatggtgcACAGTCCTCGAGGTTGACGATGG |
| YlP-SI-SOE2-F |
GTCAACCTCGAGGACTGTgcaccatccttgaatcag |
Amplification of PalI gene |
| pYLSC-SI-R |
GCTCTAGATTAATGGTGGTGGTGGTGGTGTCGC |
| 6560F |
GATCCGGCATGCACTGATC |
Colony PCR and genome identification |
| 6904R |
AACACCGGTGTTGGACTCAG |
| PMD-P1 |
GGCTTTACACTTTATGCT |
Colony PCR |
| PMD-P2 |
ACAGATGCGTAAGGAGA |
注:YlP-SI-SOE1-F斜体下划线标注部分为SfiⅠ酶切位点,YlP-SI-SOE1-R小写区域为与PalI片段重叠部分;pYLSC-SI-R斜体下划线部分为XbaⅠ酶切位点,双下划线部分为His标签编码序列;YlP-SI-SOE2-F小写区域为与Pir1片段重叠部分
Note: The underlined part of YlP-SI-SOE1-F in italics is SfiⅠ digestion site, and the lowercase area of YlP-SI-SOE1-R is the overlap with PalI fragment; The italicized underlined part of pYLSC-SI-R is the XbaⅠ digestion site, and the double underlined part is the coding sequence of His tag; The small area of YlP-SI-SOE2-F is the overlap with Pir1 fragment. |
1.5 蔗糖异构酶表面展示菌株Pir1-PalI/Po1g的构建
1.5.1 亚克隆载体pMD19-T-Pir1-PalI的构建
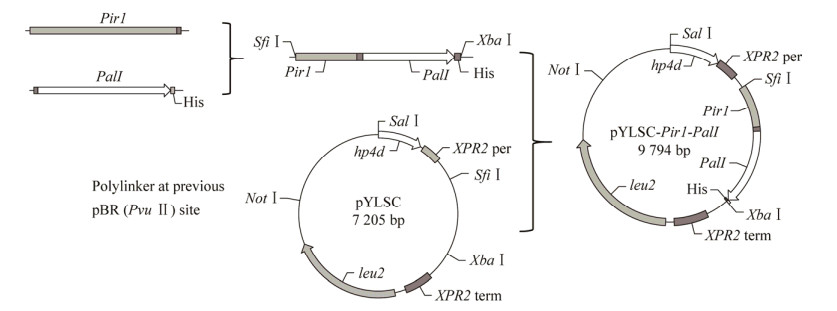
蔗糖异构酶基因PalI来自克雷伯氏菌(Klebsiella sp.) LX3,去掉该基因的起始密码子及分泌信号肽序列(大小为1 710 bp),并在其末端引入6×His标签。送至生工生物工程(上海)股份有限公司经解脂耶氏酵母密码子优化后合成基因PalI (GenBank登录号为KJ 452353.1),全基因合成后克隆至质粒pYLSC,由本实验室保存。Y. lipolytica来源的锚定蛋白Pir1基因(GenBank登录号为AF 336989),去掉起始密码子和终止子序列,由生工生物工程(上海)股份有限公司全基因合成后克隆至载体pUC57,获得含锚定蛋白Pir1基因的载体pUC57-Pir1,基因大小为861 bp。
以本实验室保存的pYLSC-PalI质粒为模板,通过PCR扩增获得编码蔗糖异构酶的基因片段PalI。以含锚定蛋白Pir1基因的载体pUC57-Pir1为模板,通过PCR扩增获得锚定蛋白编码基因片段Pir1。通过重叠延伸PCR将2个基因片段进行重叠延伸获得融合基因片段Pir1-PalI,连接方式如图 1所示。将融合基因片段Pir1-PalI亚克隆至T-vector pMD19 (simple)载体后转化感受态细胞E. coli DH10B,37 ℃静置培养16 h,挑取单菌落,利用引物PMD-P1和PMD-P2进行菌落PCR鉴定,将阳性亚克隆菌体接入LB液体培养基(含100 μg/mL氨苄西林抗生素)中,37 ℃、200 r/min培养过夜后提取质粒,将质粒送往吉林省库美生物科技有限公司测序,测序正确的克隆质粒命名为pMD19-T-Pir1-PalI。
1.5.2 蔗糖异构酶表面展示载体pYLSC-Pir1-PalI的构建
利用酶切连接的方式构建表面展示载体。首先将上述含有融合基因的亚克隆载体pMD19- T-Pir1-PalI与表面展示载体pYLSC分别进行SfiⅠ和XbaⅠ双酶切,经切胶回收后获得具有相同黏性末端的融合基因片段Pir1-PalI与表面展示载体pYLSC,利用T4 DNA连接酶进行连接,将融合基因插入到强杂合启动子hp4d和分泌信号肽(XPR2)下游,转录终止子XPR2上游(质粒构建图谱如图 1所示),然后转化入感受态细胞E. coli DH10B,37 ℃静置培养16 h,挑取单菌落,利用引物6560F和6904R进行菌落PCR鉴定,将阳性克隆菌体接入LB液体培养基(含100 μg/mL氨苄西林抗生素)中,37 ℃、200 r/min培养过夜后提取质粒,将质粒送往吉林省库美生物科技有限公司测序,将测序正确的表面展示载体命名为pYLSC-Pir1-PalI。
1.5.3 蔗糖异构酶表面展示菌株Pir1-PalI/Po1g的构建
利用位于pYLSC质粒pBR322同源序列中的NotⅠ位点对蔗糖异构酶表面展示载体pYLSC-Pir1-PalI进行线性化处理,纯化后的线型pYLSC-Pir1-PalI载体利用一步法转化入Y. lipolytica Po1g感受态细胞中,通过pBR322同源序列与Po1g基因组发生同源重组,从而整合进入基因组中。菌株Po1g为亮氨酸缺陷型,pYLSC质粒中的亮氨酸基因(leu2)可以使重组菌在缺少亮氨酸的YNB培养基中生长,因此,经30 ℃静置培养,在YNB平板中生长的菌落理论上已整合了外源基因片段。为进一步确定目的基因是否重组入Po1g基因组中,挑取单菌落接入YPD液体培养基(pH 4.0并按1:10添加0.5 mol/L柠檬酸-柠檬酸钠缓冲液)中,30 ℃、200 r/min摇床培养过夜,提取基因组DNA,利用引物6560F和6904R进行基因组PCR鉴定,以此筛选出阳性克隆菌,将鉴定正确的阳性克隆菌命名为Pir1-PalI/Po1g并保存。
1.6 表面展示的蔗糖异构酶的表达
将蔗糖异构酶表面展示菌株Pir1-PalI/Po1g进行平板活化,挑取单一菌落接种至YPD液体培养基(pH 4.0)中进行种子培养,培养基中预先按1:10加入0.5 mol/L柠檬酸-柠檬酸钠缓冲液,30 ℃、200 r/min进行摇床培养至OD600达到2.0。取1 mL种子液,按1:50接入50 mL的YPD培养基中,于30 ℃、200 r/min进行发酵。取6、12、20、24、48、72、96和120 h的发酵液,于4 ℃、8 000×g离心10 min收集菌体,以ddH2O清洗菌体2次,然后以0.5 mol/L柠檬酸-柠檬酸钠缓冲液重悬菌体,制备菌悬液。采用10% SDS-PAGE及Western blotting对菌悬液进行分析。
1.7 表面展示的蔗糖异构酶酶活力的检测
分别取24、48、72和96 h的发酵液1 mL,8 000×g离心10 min收集菌体,以ddH2O清洗菌体2次后置于105 ℃烘干箱中进行烘干,直至重量恒定,测定获得的菌体重量,即为菌体干重(dry cell weight,DCW),每组测定3个平行样。取上述不同时间的发酵液8 000×g离心10 min收集菌体,再以ddH2O清洗菌体2次后用磷酸氢二钠-柠檬酸缓冲液重悬菌体,制备菌悬液,将该菌悬液作为PalI粗酶液。利用3, 5-二硝基水杨酸(3, 5-dinitrosalicylic acid,DNS)比色定糖法测定PalI粗酶液(表面展示的蔗糖异构酶)的酶活力。
酶活力具体测定方法如下:取100 μL粗酶液(含0.15 mg干细胞),加入400 μL磷酸氢二钠-柠檬酸缓冲液(pH 6.0)配制的4%蔗糖,45 ℃反应5 min后迅速放入水浴锅中沸水浴15 min灭活终止酶反应。取150 μL反应液加入200 μL DNS,在水浴锅中煮沸5 min,反应完成后迅速取出,加入900 μL ddH2O,反复颠倒混匀后取出200 μL反应液体,测定其在540 nm处吸光值。以含空载体pYLSC的Y. lipolytica Po1g菌株pYLSC/Po1g为阴性对照,每个反应设置3个平行样。根据异麦芽酮糖标准曲线计算产生的还原糖,并根据酶活力定义计算表面展示的蔗糖异构酶酶活力。1个单位的表面展示蔗糖异构酶酶活(U)被定义为:在反应初始阶段,每分钟释放1 μmol异麦芽酮糖的干细胞的质量(DCW)。
1.8 表面展示的蔗糖异构酶酶学性质的分析
1.8.1 表面展示蔗糖异构酶的最适反应温度及热稳定性的测定
取0.15 mg发酵菌体即表面展示的蔗糖异构酶重悬于100 μL磷酸氢二钠-柠檬酸缓冲液(pH 6.0)中,分别在不同温度条件(20−60 ℃)进行反应,测定其酶活力,以确定其最适反应温度,具体酶活力测定方法见1.6;为了测定表面展示的蔗糖异构酶的热稳定性,将表面展示的蔗糖异构酶在不同温度下温育10 min后测定其剩余酶活力。
1.8.2 表面展示蔗糖异构酶的最适反应pH及pH稳定性的测定
将0.15 mg发酵菌体即表面展示的蔗糖异构酶分别置于不同pH [0.1 mol/L磷酸氢二钠-柠檬酸(pH 3.0−6.0)、0.2 mol/L磷酸氢二钠-磷酸二氢钠(pH 6.0−8.0)及0.05 mol/L Tris-HCl (pH 8.0−9.0),所有缓冲液均按照标准配方配制[19]]缓冲液中,在45 ℃条件下测定表面展示蔗糖异构酶酶活力,以测定其最适反应pH,具体酶活力测定方法见1.6;为了测定表面展示的蔗糖异构酶的pH稳定性,将表面展示的蔗糖异构酶在不同pH条件下室温温育24 h后测定其剩余酶活力。
1.9 表面展示的蔗糖异构酶转化蔗糖产物的分析
取3 mg发酵菌体即表面展示的蔗糖异构酶重悬于100 μL磷酸氢二钠-柠檬酸缓冲液(pH 6.0)中,加入400 μL的4%蔗糖溶液,反应体系为500 μL,45 ℃反应45 min。沸水15 min灭活后,8 000×g离心10 min取反应液上清,采用HPLC对转化蔗糖产物进行分析。色谱条件为:流动相乙腈: 水体积比为80:20,柱温为35 ℃,进样量为10 μL,流速为1 mL/min,洗脱时间为20 min,每个样品制备3个平行样。
2 结果与分析
2.1 蔗糖异构酶表面展示菌株Pir1-PalI/Po1g的构建结果
2.1.1 蔗糖异构酶表面展示载体pYLSC-Pir1- PalI的构建结果
通过重叠延伸PCR将锚定蛋白编码基因Pir1 (861 bp)与蔗糖异构酶编码基因PalI (末端带有6×His标签,片段大小为1 728 bp)进行融合,获得大小为2 589 bp的融合基因片段Pir1-PalI,产物经琼脂糖凝胶电泳检测,测定结果与预期条带大小一致,融合基因扩增成功。使用胶回收试剂盒回收Pir1-PalI基因片段,将该片段亚克隆至载体T-vector pMD19 (simple),转化至大肠杆菌E. coli DH10B感受态细胞中,37 ℃静置培养16 h,挑取单菌落进行菌落PCR鉴定。目的条带理论大小为2 978 bp,菌落PCR产物经核酸电泳验证,条带大小与预期大小符合。将阳性克隆菌体接入LB液体培养基中37 ℃、200 r/min培养过夜,提取质粒送往吉林省库美生物科技有限公司测序,测序结果显示与目的基因序列完全一致,说明亚克隆载体构建成功,将其命名为pMD19-T-Pir1-PalI。将亚克隆载体pMD19-T-Pir1-PalI与表面展示载体pYLSC分别进行SfiⅠ和XbaⅠ双酶切,利用T4 DNA连接酶将2个含有相同黏性末端的片段进行连接,然后转化到E. coli DH10B感受态细胞中,并于LB (Amp抗性)平板37 ℃静置培养过夜,随机挑取平板上的10个单菌落进行菌落PCR鉴定。目的条带理论大小为2 978 bp,菌落PCR产物经核酸电泳验证,条带大小与预期大小符合。提取菌落PCR鉴定正确的克隆菌株的质粒,并将其送往吉林省库美生物科技有限公司测序,测序结果显示,其与目的基因序列完全一致,说明成功构建了表面展示载体,将其命名为pYLSC-Pir1-PalI。
2.1.2 蔗糖异构酶表面展示菌株Pir1-PalⅠ/Po1g的构建结果
蔗糖异构酶表面展示载体pYLSC-Pir1-PalI经NotⅠ线性化处理后转入Y. lipolytica Po1g感受态细胞中,从YNB平板上挑取6个单菌落接入YPD液体培养基,于30 ℃、200 r/min培养18−20 h后,提取基因组DNA进行PCR验证。基因组PCR产物经1%琼脂糖凝胶电泳分析。目的条带理论大小为2 978 bp,PCR产物经核酸电泳验证,条带大小与预期大小相符合。说明成功获得Y. lipolytica表面展示蔗糖异构酶重组菌株,命名为Pir1-PalI/Po1g。
2.2 表面展示的蔗糖异构酶的表达结果
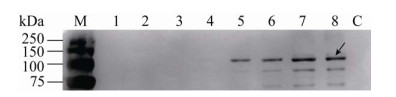
将成功构建的蔗糖异构酶表面展示菌株Pir1-PalI/Po1g接入发酵液进行30 ℃、200 r/min摇床培养发酵,对发酵液进行6、12、20、24、48、72、96和120 h定时取样,通过10% SDS-PAGE及Western blotting测定上述培养时间下蔗糖异构酶在Y. lipolytica表面的展示情况。SDS-PAGE测定结果如图 2所示,图 2中黑色箭头所示条带大小在目的蛋白99.20 kDa的理论值大小范围内,随着培养时间的延长,目的蛋白的表达量增加,直到72 h达到最大,继续发酵目的蛋白的表达量无明显变化。Western blotting测定结果如图 3所示,图 3中黑色箭头所示条带大小在目的蛋白99.20 kDa的理论值大小范围内,这与SDS-PAGE的测定结果一致,说明蔗糖异构酶在Y. lipolytica表面成功展示。
2.3 表面展示的蔗糖异构酶酶活力的测定结果
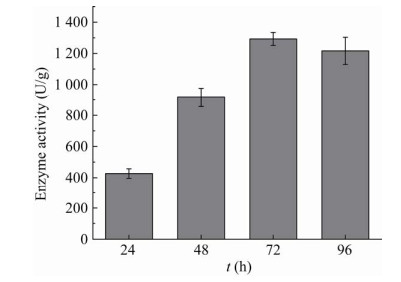
将成功构建的蔗糖异构酶表面展示菌株Pir1-PalI/Po1g接入发酵液进行30 ℃、200 r/min摇床培养发酵,对发酵液进行定时取样,测定24、48、72和96 h发酵菌体即表面展示的蔗糖异构酶酶活力,测定结果如图 4所示。随着培养时间的增加,蔗糖异构酶表面展示菌体酶活力逐渐增大直至平稳,而且最大酶活力出现在72 h,达到1 291.81 U/g-DCW,继续发酵酶活力值不发生变化,后续选取72 h发酵菌体进行酶活力测定。
2.4 表面展示的蔗糖异构酶的酶学性质的分析
2.4.1 表面展示蔗糖异构酶的最适反应温度及温度稳定性的测定结果
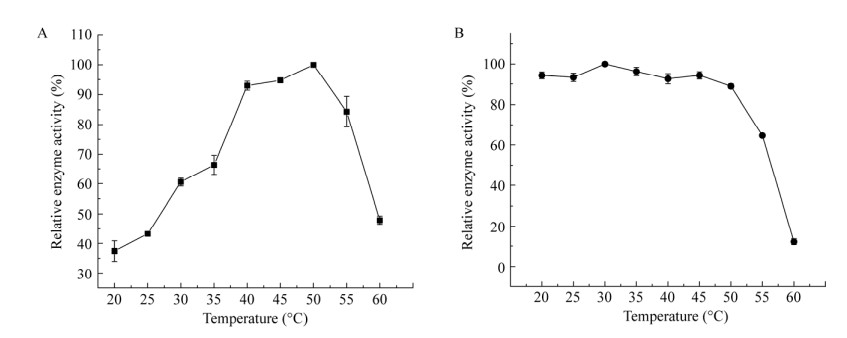
以4%蔗糖为底物,在不同温度条件下测定表面展示蔗糖异构酶酶活力。测定结果如图 5A所示,当反应温度达到50 ℃时,酶活力值达到最高,并且当温度处于40−45 ℃时,酶活力无明显变化。然而随着温度的继续升高,酶活力逐渐降低,在60 ℃时几乎完全失活。将该酶在不同温度下保温10 min后测定其剩余酶活力,测定结果如图 5B所示,在20−45 ℃孵育10 min后,剩余酶活力仍保留85%以上;当温度升至55 ℃时酶活力迅速下降,但依然能保持在60%;当温度进一步升高到60 ℃时已经很难检测到活性,这说明表面展示蔗糖异构酶在55 ℃以内有良好的稳定性。
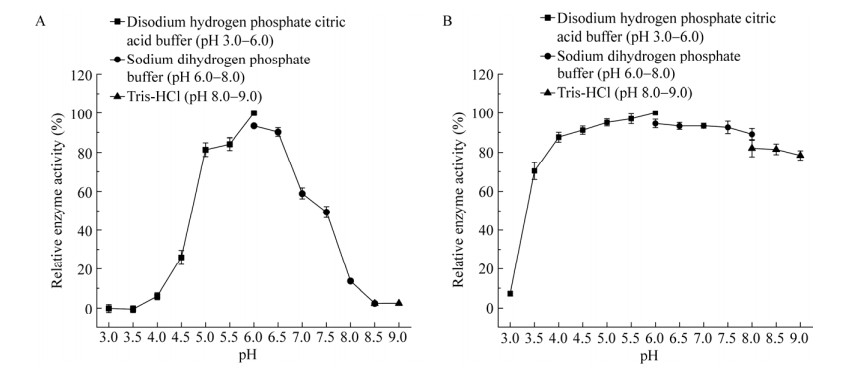
2.4.2 表面展示蔗糖异构酶的最适反应pH及pH稳定性的测定结果
以4%蔗糖为底物,在不同pH条件下测定表面展示蔗糖异构酶酶活力。测定结果如图 6A所示,最适反应pH值为6.0,pH值小于4.5或者大于8.0时,酶活力大幅度降低。将该酶在不同pH下室温孵育24 h后测定其pH稳定性,测定结果如图 6B所示。在pH 4.5−9.0孵育24 h后,剩余酶活力仍保留80%以上;在pH值小于4.0时,酶活力迅速下降;pH值为3.0时已经很难检测到活性,说明该酶具有较宽的pH稳定区间。
2.5 表面展示蔗糖异构酶转化蔗糖产物的分析
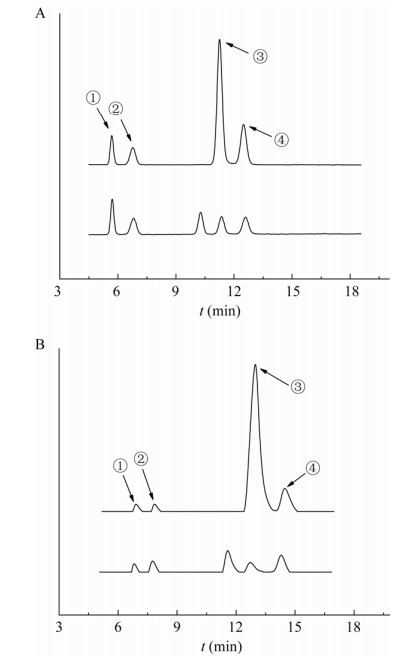
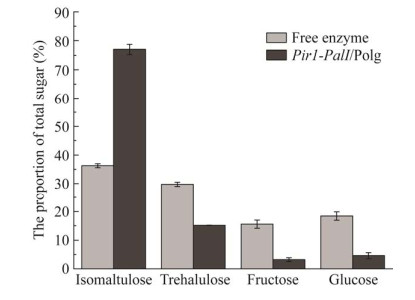
通过HPLC分析蔗糖异构酶转化蔗糖的产物中各组分占比,以在Y. lipolytica Po1g中外泌表达游离的蔗糖异构酶为阴性对照。测定结果如图 7所示,蔗糖转化产物主要是异麦芽酮糖和海藻酮糖,但伴随单糖副产物葡萄糖及果糖的生成。根据标准品(1%异麦芽酮糖,1%海藻酮糖,1%葡萄糖,1%果糖)的出峰时间确定样品中的各个组分的出峰时间。分别取浓度为0.01%−5.00%的标准品进行HPLC,以标准品浓度为横坐标、以峰面积为纵坐标绘制不同标准品的标准曲线,将转化产物中的不同组分的峰面积代入标准曲线,计算得出转化产物中各组分的具体生成量,并计算其在总产物中的占比,测定结果如图 8所示。与游离蔗糖异构酶相比,表面展示的蔗糖异构酶转化蔗糖产物中异麦芽酮糖的含量由36.29%提高到了77.01%±1.77%,为(13.86±0.08) mmol/L;而海藻酮糖、果糖及葡萄糖在产物中的占比较低,分别为15.17%±0.06%、3.21%±0.67%及4.61%±1.03%。这说明展示于Y. lipolytica Po1g表面的蔗糖异构酶转化蔗糖产物中的单糖比例较游离的酶有了明显的降低。
3 讨论
本研究构建了一个蔗糖异构酶表面展示系统,该系统中展示于Y. lipolytica Po1g表面的蔗糖异构酶转化蔗糖的产物异麦芽酮糖的含量较该酶的游离状态下有显著提高,异麦芽酮糖含量由36.29%提高至77.01%±1.77%。这种产物比例的差别可能是由于酶活性位点附近空间结构的变化造成的。以往的报道表明,利用Pir1在S. cerevisiae表面展示糖基转移酶的展示系统中,酶的分布不规则[17]。因此,本研究采用的展示系统可能是由于锚定蛋白Pir1在细胞壁上的不规则分布,导致与其共表达的蔗糖异构酶在细胞壁上的不规则分布,直接影响到其与底物的作用[20],最终导致表面展示的蔗糖异构酶转化蔗糖的产物中各个组分的占比发生变化。另外,本研究中蔗糖异构酶在其来源菌中表达时,其酶的产物组成比例与酶的表达场所有关,其中以胞壁酶催化形成的葡萄糖与果糖产物比例最低[20]。因此,将其进行表面展示有助于降低其单糖产物的形成,与上述因素共同作用改变了蔗糖异构酶转化产物的比例。
本研究中展示在Y. lipolytica Po1g表面的蔗糖异构酶与其他表达菌株中表达的重组蔗糖异构酶相比,蔗糖异构酶的温度耐受性及pH耐受性都得到了提高,在pH 4.5−9.0条件下孵育24 h后剩余酶活力仍保留80%,并且能在20−55 ℃环境下保持酶活力相对稳定。来自Erwinia sp.的蔗糖异构酶在pH 5.0−7.0及温度处于30 ℃以下时才较稳定[3];Erwinia sp. D12产生的蔗糖异构酶在pH 5.7−6.3及5 ℃条件下只能在24 h内保持酶活力稳定[21];Klebsiella sp. 18中纯化得到的蔗糖异构酶在相同的测定条件和pH 5.5−6.6范围内稳定[22]。稳定性的提高可能是由于展示的酶被锚定在细胞膜上,而细胞膜稳定了酶的结构,防止其被蛋白酶水解,从而获得了较高的pH稳定性及温度稳定性。
4 结论
本研究成功构建了蔗糖异构酶的Y. lipolytica Po1g表面展示菌株Pir1-PalI/Po1g,实现了蔗糖异构酶在Y. lipolytica细胞表面的固定化。经测定,表面展示的蔗糖异构酶的最高酶活力为(4 694.6±56.6) U/g-DCW,最适反应条件为45 ℃和pH 6.0,在20−55 ℃及pH 3.5−9.0环境下酶活力稳定,较其他表面展示系统展示的蔗糖异构酶而言,温度及pH稳定性显著提高。转化蔗糖获得异麦芽酮糖的转化率为77.01%±1.77%,较分泌表达的蔗糖异构酶及其他表面展示系统展示的蔗糖异构酶而言,该表面展示的蔗糖异构酶获得了更高的异麦芽酮糖得率及更低的单糖得率。
 2022, Vol. 49
2022, Vol. 49